Crystallizing science, one protein at a time
It’s square one. It’s step one. It puts the “basic” in basic science. How ever you describe it, understanding protein structure and function through what’s called X-ray crystallography is an important approach in many areas of biochemistry, including drug design. And it’s a technique many researchers in the Department of Biochemistry at the University of Wisconsin–Madison specialize in.

A computer repairperson can’t fix or improve a computer without knowing anything about the parts, and a scientist can’t work with a protein properly without knowledge of its basic structure first. If a mutation in a protein causes disease, knowing where the mutation is located — and its effects there — could help find a way to fight the disease. Oftentimes, structure holds the key.
The process required to determine, or “solve,” the structure of a protein through X-ray crystallography has changed rapidly, like many scientific techniques. It’s gone from long and complex to an approachable technique that is used by many researchers.
In crystallography, the protein of interest has to be purified and coaxed into crystallizing. Next the crystal is exposed to X-rays, which are scattered by the crystal, and data are collected on these scattered X-rays. Mathematical equations and computer programs take it from there, helping the researcher generate a three-dimensional image of the protein. This process once took decades. As science advanced it began to take five years, then one, and today, if all goes well, just a matter of weeks.

“Our main goal is to be able to see what a protein or other molecule looks like in three dimensions,” says Hazel Holden, a professor in the UW–Madison Department of Biochemistry. “In the case of enzymes, we want to understand how they catalyze their reactions, which can only be fully understood by a three-dimensional picture.”
Holden’s focus is on enzymes, while a fellow faculty member in Biochemistry, Ivan Rayment, is interested in motor proteins, which use energy to move necessary components around in a cell. While he is interested in many other types of molecules, his work on motor proteins began with the study of myosin, the major protein in muscle, solving the first structure of it in the early 1990s.
Myosin converts chemical energy in muscle to physical energy for directed movement. The structure took him and his colleagues 11 years to solve back then.

“We don’t just solve structures to make pretty pictures,” he says. “We solve structures to understand what’s going on at the molecular level. The crystal gives you ideas about atoms, and atoms make the difference. One mutated amino acid in a protein can be the difference between life and death. Often that’s just three or four atoms. They control your destiny, so details are important.”
Crystallization: The Process
Proteins differ in size, shape, polarity, and more — all influenced by the number and arrangement of atoms of different elements like carbon, oxygen, and nitrogen. This number and arrangement determine how the protein folds and ultimately functions.
To begin to solve a protein structure, it needs to be crystallized. In a protein crystal, millions of individual proteins are arranged in the same orientation in a crystalline lattice. Think of something analogous to individual salt crystals, which are collections of sodium and chlorine ions.

Getting the proteins to make this crystalline lattice is tricky but has also gotten much easier over the years. It differs for each protein because each protein is unique. So, researchers try many different conditions, adding different chemicals, changing the pH or temperature, even changing tiny parts of the protein — in the hopes the proteins will line up perfectly in the same orientation into a crystalline lattice.
Crystallographers employ several different methods to achieve this. One, called vapor diffusion, was actually invented by Bob Bock, a former biochemistry faculty member at UW–Madison and dean of the Graduate School. The method consists of mixing a drop of liquid containing the protein of interest with another drop containing the so-called precipitant, the agent that helps the protein to crystallize. The mixture is then placed in close proximity to a larger volume of the precipitant so that over time, the concentration of the precipitant in the protein mixture increases which occasionally results in crystallization.
“Sometimes crystals appear in the time it takes you to get a cup of coffee,” Holden explains. “Often, however, it takes days or weeks or, to the dismay of my graduate students, months. And you have to get a decent sized crystal to get good data.”
Visualizing Proteins
Obtaining a good crystal is only half the battle. The next step requires a large piece of equipment to shoot X-rays at the crystal. This is sometimes an in-house X-ray generator, but more often a synchrotron facility such as that located at Argonne National Labs in Illinois. Researchers expose the crystal to the X-rays by suspending it in a tiny amount of frozen liquid. The X-rays hit the crystal and scatter in all directions and are captured by a detector. This scattered radiation, along with other data the researchers utilize, are used to produce an electron density map. This map can be likened to a “road map,” telling the investigator what the protein looks like in three-dimensional space. The investigators use this map to build the protein model.
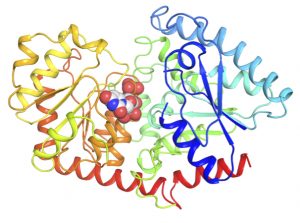
In recent years, technology has changed the game when it comes to visualizing the protein models generated from X-ray crystallographic analyses. Computer software packages such as PyMOL allow these protein models to be visualized even by students who do not work in the field.
“It’s a night and day difference between how we used to make crystals and assemble structures and how we do it now,” Rayment says. “Technology has changed a lot and now x-ray crystallography can be incorporated into the research of many students and faculty. As we’ve improved the technology, the focus has become the biology of the molecules. We are now able to put more effort into uncovering the biological function behind the structure.”
Donning 3D glasses, researchers see each bond come to life on their screen as they explore their electron density maps. Each atom and carbon-carbon bond science students draw time and time again in biochemistry classes has a real and tangible purpose. A structure can be assembled in an afternoon or a week, a sharp contrast to the five years it took Holden to complete the model that earned her a Ph.D. in ’82 with an entire room full of computers and hand models. In fact, Holden teaches middle school students how to perform these techniques in her outreach program titled Project CRYSTAL — Colleagues Researching with Young Scientists, Teaching and Learning.
“What’s the Point?”
“Now a lot of people, including my parents, used to ask ‘what’s the point?’ ” Holden says. “Well we solve the structures, which is the basic science component, and others look at our work and use the structures in various areas of science, including drug design.”
For example, in the Rayment Laboratory, the team has worked on toxins produced by fungus that harm crop yields. He’s also been involved in work on cardiac myosin in the heart that could have implications for heart disease.
Other work in structural biology, not from a UW–Madison laboratory, has helped lead to treatments for breast cancer, HIV, and high blood pressure. In Holden’s laboratory, she investigates enzymes that are involved in the synthesis of complex and unusual sugars which appear on the outside of the cells of some bacteria. She believes some of these sugars may be what help the bacteria cause infection. Understanding the structures of the enzymes that make these sugars may help to find a way to prevent the bacteria from making them — effectively serving as an antibiotic.
Rayment adds that a very important part of this field’s structural biology work is that information on newly solved structures is uploaded to a database and made freely available. That wasn’t always the case though, as scientists used to be more secretive about a protein that took them 10 years to solve. But as the process sped up it became more important to share this taxpayer-funded work for the benefit of all.
“I hope the work that I do stands the test of time and that I make contributions which are helpful to others and we advance our understanding of structure and function,” he says. “The beauty of X-ray crystallography or any high-resolution imaging technique is that it gives you a special representation of where atoms are so you can generate a more robust hypothesis of how something works. And it’s just a huge amount of fun.”
This story was originally published on the Department of Biochemistry News site.